











.png)


9 de junio, 2020
12 minutos
En Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios en España, podemos encontrar la definición de producto sanitario, medical device en inglés:
«Producto sanitario»: cualquier instrumento, dispositivo, equipo, programa informático, material u otro artículo, utilizado solo o en combinación, incluidos los programas informáticos destinados por su fabricante a finalidades específicas de diagnóstico y/o terapia y que intervengan en su buen funcionamiento, destinado por el fabricante a ser utilizado en seres humanos con fines de:
- 1.º Diagnóstico, prevención, control, tratamiento o alivio de una enfermedad,
- 2.º diagnóstico, control, tratamiento, alivio o compensación de una lesión o de una deficiencia,
- 3.º investigación, sustitución o modificación de la anatomía o de un proceso fisiológico,
- 4.º regulación de la concepción, y que no ejerza la acción principal que se desee obtener en el interior o en la superficie del cuerpo humano por medios farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales medios.
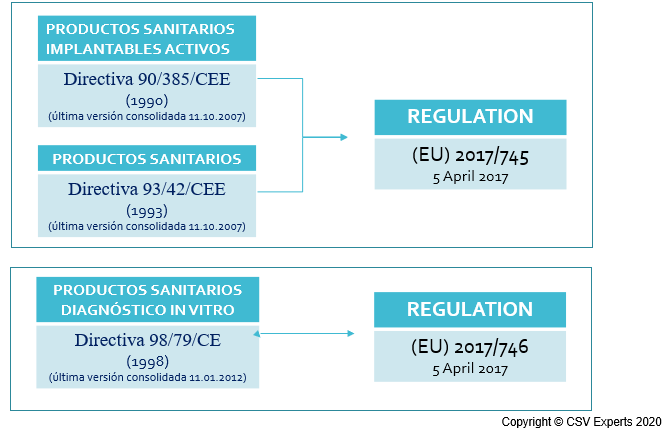
Actualmente la normativa que aplica a los productos sanitarios se encuentra en “período de transición”, es decir, de cambio hacia un nuevo reglamento, el MDR 2017/745/UE, que ha retrasado su fecha de aplicación un año.
Tenía que entrar en vigor el 26 de mayo de 2020, y debido al desabastecimiento de muchos productos sanitarios y la implicación que se necesita por parte de las autoridades sanitarias, como resultado de la pandemia por COVID-19, se ha retrasado un año.
Su nueva fecha de aplicación es el 26 de mayo de 2021, dejando así un poco más de margen a las autoridades competentes y organismos notificados para que puedan concentrarse en que aquellos productos de los que hay más demanda puedan llegar al mercado.
Regulación de los dispositivos médicos en la Unión Europea
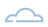
Como ya se ha introducido, estamos en período de transición y las directivas europeas que están en vigor para productos sanitarios son las que se muestran en la figura siguiente, con 3 directivas:
- 90/385/CEE de productos sanitarios implantables activos
- 93/42/CEE de productos sanitarios
- 98/79/CE de productos sanitarios de diagnóstico in vitro
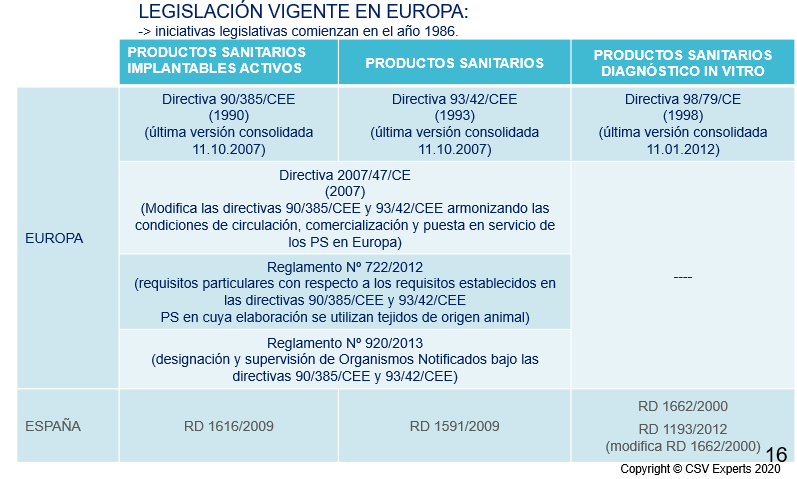
De estas 3 directivas, que necesitan transposición en cada uno de los países miembros en sus respectivas normas, leyes, órdenes, o reales decretos entre otros, vamos a pasar a dos reglamentos que aplicarán por igual a todos los países de UE.
De esta manera se busca que en todos los países apliquen la normativa con el mismo rigor, o evitar diferencias, ventajosas o no, entre los diferentes estados miembros.
 Los nuevos reglamentos europeos de producto sanitario buscan aumentar la seguridad del paciente y del servicio de salud pública en general, en los países miembros de la Unión Europea.
Los nuevos reglamentos europeos de producto sanitario buscan aumentar la seguridad del paciente y del servicio de salud pública en general, en los países miembros de la Unión Europea.
En este artículo vamos a hablar de los productos sanitarios de acuerdo al nuevo reglamento o MDR, sin tener en cuenta los productos de diagnóstico in vitro, cuyas características específicas tiene su propio reglamento (IVDR 2017/746/EU), y cuya fecha de aplicación es el 26 de mayo de 2022.
MDR introduce grandes cambios lo que supone un reto para todos los agentes económicos implicados en la fabricación, importación y distribución de los productos sanitarios o medical devices. De manera resumida, habrá variaciones en los siguientes aspectos y sentidos:
- Ha cambiado la clasificación, pasando a ser mayor, de numerosos productos sanitarios, lo que implica mayores requisitos a nivel regulatorio.
- Se van a requerir un mayor número de datos clínicos e investigaciones clínicas, sobre todo para los productos sanitarios de mayor riesgo (Clase IIb implantables y Clase III).
- Se endurecen los requisitos postcomercialización, donde se exige a todos los agentes económicos una colaboración en la recopilación, muy proactiva de datos clínicos y otros, sobre el comportamiento de los productos sanitarios una vez llegan al mercado.
- Mayores requisitos de trazabilidad de los productos, incluyendo UDI (Unique Device Identification), un número de identificación única para todos los productos sanitarios.
- EUDAMED: una nueva base de datos, a la que se accede mediante un número UDI básico, donde se registrarán los productos, los fabricantes, representantes autorizados (para productos fabricados fuera de la UE) e importadores y que será una herramienta de control y vigilancia de mercado.
- Se exige una tarjeta para cada uno de los productos implantables.
- Los fabricantes deberán tener un seguro de responsabilidad civil que cubra productos defectuosos y el daño que éstos puedan ocasionar.
- Se exige una figura: la de persona responsable del cumplimiento de la normativa, en las empresas fabricantes y también en las de representantes autorizados.
Así mismo, el nuevo reglamento tiene un alcance mayor, al afectar también a productos sin finalidad médica, pero que tienen el mismo mecanismo de acción que equivalentes en el mundo del producto sanitario, como es el caso de muchos productos cosméticos como láseres o equipos de liposucción. Para estos equipos o productos la comisión publicará unas especificaciones comunes de obligado complimiento.
Otro de los puntos en los que más impactan los nuevos reglamentos es en la vigilancia y control sobre los Organismos Notificados que deberán pasar por procesos de recertificación para poder adaptarse. Los Organismos Notificados son organizaciones designadas por los países de la UE que evalúan la conformidad de ciertos productos antes de ser comercializados.
Estos organismos llevan a cabo tareas relacionadas con los procedimientos de evaluación de la conformidad establecidos en la legislación aplicable, cuando se requiere que éstos sean supervisados por un tercero. La Comisión Europea publica en la base de datos Nando una lista de dichos organismos notificados.
Los cambios a nivel regulatorio obedecen por una parte a la necesidad de adaptase a la innovación y nuevas tecnologías que quedaban cubiertas de una manera muy laxa con las directivas, y el otro motivo, y gran desencadenante del endurecimiento legislativo en Europa fue el fraude de la empresa PIP (Poly Implant Prothèse), que estaba fabricando implantes de silicona destinados a ser usados en prótesis mamarias elaborándolos con silicona no quirúrgica.
Como consecuencia, comenzaron a reportarse casos de roturas, e incluso algunas de las pacientes terminaron desarrollando cánceres entre otros muchos efectos adversos. Este caso tan vergonzoso y lamentable llevó a la comisión europea a crear el llamado “Plan de Acción PIP”, en el que se comenzaron a revisar aspectos relativos al funcionamiento de los organismos notificados, a la vigilancia post-comercialización, a la coordinación de organismos notificados y a las autoridades competentes para aumentar la seguridad de los pacientes.
La clasificación de los dispositivos médicos
La nueva regulación europea, al igual que la directiva MDD vigente, clasifica los productos sanitarios en 4 clases en función del riesgo:
Clase I, IIa, IIb y III, también hay un grupo especial de Clase I: los estériles, los de medición y los quirúrgicos reutilizables (éstos últimos son una novedad).
Exceptuando los de Clase I, todos los demás productos, incluyendo los Clase I estériles, de medición y quirúrgicos reutilizables, necesitan la intervención de un organismo notificado, es decir necesitan la participación de esa tercera parte que supervisa que los productos son conformes respecto a la legislación de la UE aplicable.
 Situación actual
Situación actual
La Comisión Europea es consciente de que el impacto de la nueva normativa en las organizaciones es considerable, y requiere de un aumento de recursos destinados a asegurar la conformidad con la nueva regulación.
Para permitir una transición más suave y no desabastecer el mercado, el nuevo reglamento entrará en vigor paulatinamente: aquellos productos sanitarios que, por su clase de riesgo, necesiten de un organismo notificado para obtener su marcado CE, podrán tener sus productos en el mercado mientras tengan un certificado CE vigente contra MDD, como máximo hasta el año 2024, y un año más, 2025 en distribución.
Por supuesto, los productos sanitarios que continúen en el mercado según MDD no podrán tener modificaciones de diseño durante ese período, y deberán cumplir de todas formas con las obligaciones de UDI, registro de agentes económicos en EUDAMED, en cuanto entre en vigor, en mayo de 2022, y seguimiento post-comercialización según Reglamento MDR.
 Conclusiones
Conclusiones
- Los fabricantes y todos los agentes económicos implicados en la fabricación o puesta en el mercado de cualquier producto sanitario tienen que adaptarse a los nuevos reglamentos de productos sanitarios.
- El retraso de un año debido a la pandemia COVID-19 es una buena noticia, pero no es la panacea, ya que sigue habiendo muy poco tiempo de preparación, teniendo en cuenta la cantidad de nuevos datos e informes que se requieren.
- Los fabricantes deben corroborar que disponen de suficientes datos clínicos para asegurar la seguridad y eficacia de sus productos y que tienen la documentación y sistemas de calidad necesarios en su organización.
- Hay aún muy pocos organismos notificados preparados para MDR, lo que supone un claro cuello de botella y puede suponer que muchos productos se queden fuera del mercado.
AMBIT BST
En AMBIT somos expertos desde hace más de 15 años en el desarrollo de estrategias y soluciones IT para tu compañía. Somos consultores e integradores en múltiples ámbitos y si quieres conocer más de las soluciones que te podemos ofrecer en el campo de la salud, no dudes en ponerte en contacto con nosotros.








.png)
Cuéntanos tu opinión