











.png)


15 de abril, 2021
7 minutos
IVDR son las siglas de una regulación europea, la Regulación sobre Productos Sanitarios para Diagnósticos in Vitro también (EU) 2017/746.
El reglamento cuya fecha de aplicación es el 26 de mayo de 2022, y que está en vigor desde 2017, se ocupa de regular la comercialización y puesta en servicio de los productos sanitarios, o en inglés medical devices que componen el grupo de los productos sanitarios para el diagnóstico in vitro en todo el territorio de la Unión Europea, así como Irlanda del norte tras el Brexit.
Plazos para IVDR
La Regulación (UE) 2017/746 se publicó el 5 de mayo de 2017 y entró en vigor el 26 de mayo del mismo año, con un plazo de cinco años de transición desde tal fecha para ser de obligado cumplimiento. Derogará a la directiva 98/79/CE sobre productos sanitarios para diagnóstico in vitro aún en vigor
Por lo que se refiere a los fabricantes de este tipo de productos sanitarios para diagnóstico in vitro, deben actuar ya, si no han comenzado su adaptación, para poder cumplir con la legislación cuando sea de obligado cumplimiento en el 2022, ya que como veremos, el nuevo reglamento introduce grandes cambios y supone un gran impacto para la industria. IVDR iguala en exigencia al reglamento de Producto sanitario (EU) 2017/745 que aplicará obligatoriamente desde el 26 de mayo de 2021.
Para permitir una transición a reglamento más suave, y evitar el desabastecimiento de productos sanitarios de diagnóstico in vitro la comisión ha previsto un período de gracia, al que los fabricantes pueden acogerse si cumplen una serie de condiciones. Así pues, se podrá fabricar y comercializar un producto certificado bajo directiva 98/79/CE dos años más a partir del 26 de mayo de 2022. Como adelantamos, a este período de gracia se podrán acoger los productos que cumplan una serie de condiciones:
- Aquellos productos que con la directiva hayan sido de autocertificación, (en los que el fabricante se marcaba el producto CE y emitía la CE-Declaración de conformidad, sin la intervención de un Organismo Notificado), y con el reglamento lo vayan a necesitar. Es decir, será de aplicación a los productos que con el reglamento vayan a ser de Clases B, C y D;
- y además, no tengan cambios en el diseño del producto;
- cumplan con el registro de actores en Eudamed, o autoridades competentes mientras éste no sea funcional;
- cumplan seguimiento poscomercialización y vigilancia, tal y como lo pide IVDR.
Además, se permitirá un año más de distribución del producto bajo directiva, hasta 25 de mayo de 2025, pero entre 26 de mayo de 2024 y 26 de mayo de 2025 ya no se permitirá fabricación bajo certificación de la directiva IVDD.
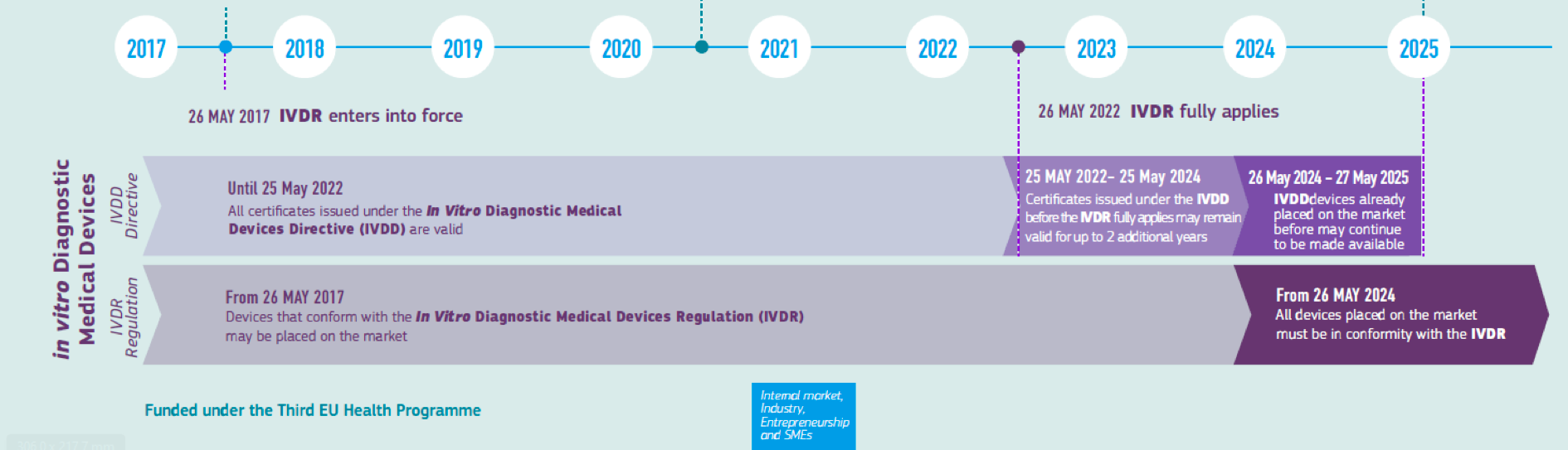
Fuente: Comisión Europea https://ec.europa.eu/docsroom/documents/34907
Introducción de este tipo de productos sanitarios en el mercado
Según se recoge en el artículo 110 del reglamento de Productos sanitarios para diagnóstico in vitro (RPSDIV), es posible introducir en el mercado nuevos artículos que cumplan con la nueva regulación antes de la fecha del 26 de mayo del 2022. Por ello y porque esta legislación es compleja y obliga a llevar a cabo cambios en el sistema de calidad y expediente técnico de los productos para obtener el marcado CE de producto sanitario, los fabricantes deberían estar ya estudiando cómo van a realizar los cambios exigidos.
En el artículo 110, apartado 5 de la IVDR se explica también que la posibilidad de introducir los productos afectados por la nueva normativa en el mercado antes de expirar el plazo dado para su adaptación, siempre y cuando cumplan todos los requisitos exigidos, es aplicable a cualquier producto sanitario para el diagnóstico in vitro, con independencia de su clase de riesgo. Hay que recordar que una de las clasificaciones de los dispositivos médicos o medical devices se basa, precisamente, en el nivel de riesgo que implica el uso de los mismos.
En el caso de productos de Clase D, los de mayor riesgo, están obligatoriamente sujetos, para la evaluación de conformidad y obtención de marcado CE a análisis del producto en laboratorios de referencia asignados por la comisión.
Grandes cambios que introduce IVDR
Como ya adelantábamos, IVDR introduce cambios de gran impacto, vamos a ver cuáles:
- Cambio radical en la clasificación de los productos. Se clasifican en base a riesgo, y de dos listas (A,B) y resto que eran autocertificados pasamos a Clases: A, B, C y D. Siendo A la de menor riesgo y que no necesita ON (Organismo Notificado), mientras que las demás sí.
- Vamos a pasar de un 80-85% de productos autocertificados a un 85% que van a necesitar ON, sólo un 20% van a ser autocertificados con reglamento.
- Un mayor alcance: se cubre la venta por internet, y los laboratorios que desarrollan los llamados test “in-house” tienen que cumplir al menos los requisitos de Anexo I, así como algunas disposiciones.
- Mayor escrutinio a los productos de mayor riesgo: Clase D necesitará pasar análisis por laboratorio de referencia designado por la comisión y los Clase C - Companion Diagnostics (Pruebas diagnósticas de selección terapéutica) van a tener que consultar con las autoridades competentes.
- Se requiere una Persona Responsable del Cumplimiento de la Normativa (PRRC), que tiene que cumplir con una serie de requisitos.
- Aumento exponencial de los requisitos de documentación: del sistema general de calidad (Quality Management System-QMS) basado en ISO 13485; de la documentación técnica, con una mayor exigencia en riesgos (ISO 14971), y documentación de evaluación de funcionamiento (Validez científica, analítica y clínica).
- Mayores requisitos de seguridad y seguimiento poscomercialización, más informes con frecuencias establecidas según la clase de riesgo del producto (PMS, PSUR, PMPF, SSCP).
- Mayores requisitos de trazabilidad: obligación de asignar un UDI (Basic UDI-DI, UDI-DI, UDI-PI).
- Eudamed, que entrará en Vigor junto con el reglamento IVDR el 26 de mayo de 2022, y que irá poniendo a disposición sus módulos paulatinamente. El módulo de registro de actores ya es funcional desde diciembre de 2020.
La comisión podrá publicar especificaciones comunes, para aquellos productos que no dispongan de normas armonizadas o guías de buenas prácticas establecidas.
Seguro de responsabilidad civil que cubra defectos en los productos sanitarios y posibles daños a usuarios o pacientes.
También se prevén Paneles de expertos, MDCG (Medical Devices Coordination Group) que se podrán consultar (productos de alto riesgo y autoridades competentes) y actos de ejecución de la comisión que cambien o aclaren determinadas normas.
Medidas de adaptación: ¿Cómo prepararnos?
Los cambios son sustanciales, así que debemos conocer cuál es la situación de la empresa, analizarla y poner en marcha las acciones necesarias para cubrir todos los requisitos de la nueva regulación.
Proponemos comenzar por un análisis de desviaciones, un análisis de GAPs, que nos permita conocer nuestras carencias y el establecimiento de un plan de acción, con acciones de formación, generación de nueva documentación que nos permitan pasar con éxito una auditoría del sistema de calidad, así como de la documentación técnica.
Punto muy crítico: disponibilidad de Organismos Notificados (ON)
Uno de los puntos más críticos va a ser la disponibilidad de organismos notificados, que vamos a poder comprobar en la base de datos NANDO (EUROPA - European Commission - Growth - Regulatory policy - NANDO)
Al contrario de lo que sucede con la directiva IVDD, en la que un 80% de los productos son autocertificados, y un 20% necesitan ON, este porcentaje se va a invertir con el reglamento de productos sanitarios para diagnóstico in vitro. Es decir, se calcula que serán un 80-85% los que van a necesitar ON y de momento tenemos sólo 4 ON disponibles para IVDR, así que si nuestra empresa es una de las que los necesita es un punto muy estratégico el hacerse
¿Qué sucederá después del 26 de mayo del 2022?
Los productos que puedan beneficiarse del período de gracia de dos años hasta el 26 de mayo de 2024, podrán seguir comercializándose o poniéndose a disposición en el mercado, siempre que no haya cambios en el diseño y se cumplan las disposiciones de registros de actores y productos, PMS (Seguimiento poscomercialización) y Vigilancia. Es decir, tendremos algún tiempo más para adaptarnos.
Para los productos que sean de Clase A con IVDR aplicará desde el 26 de mayo de 2022, así que los fabricantes de esos productos no van a poder beneficiarse del período de gracia y tendrán un período más corto de adaptación.
En el caso de productos nuevos, que salgan al mercado después a partir del 26 de mayo de 2022 ya les aplicará IVDR directamente. Desde Ambit-BST ofrecemos servicios de consultoría en producto sanitario para ayudar a las empresas a adaptarse a IVDR. No dudéis en consultarnos.
AMBIT BST
En AMBIT somos expertos desde hace más de 15 años en el desarrollo de estrategias y soluciones IT para tu compañía. Somos consultores e integradores en múltiples ámbitos, y si quieres conocer más de las soluciones que te podemos ofrecer, no dudes en ponerte en contacto con nosotros.
En Ambit BST, podemos ayudarte a garantizar la integridad de tus datos y optimizar tus procesos. Contáctanos y te ayudaremos con todo lo necesario.








.png)
Cuéntanos tu opinión