











.png)


19 de mayo, 2020
11 minutos
La cualificación de los equipos de producción es necesaria para las industrias farmacéuticas, clínicas y de dispositivos médicos, altamente reguladas, ya que si surgen inconsistencias en los mismos podrían desencadenar incumplimientos en las regulaciones e incluso la retirada del mercado de los productos fabricados con dichos equipos.
El primer paso para llevar a cabo la cualificación de los equipos de producción en el sector farmacéutico y de la salud, será evaluar el impacto del equipo en función de su criticidad GxP. Eso es, la aplicación de las normativas de buenas prácticas dentro de la industria del sector regulado para asegurar que los suministros tengan un nivel de calidad aceptable y una seguridad al paciente o consumidor.
Por lo tanto, se deberán definir los procesos críticos dentro del sistema, empezando por los de mayor criticidad, definir los flujos de datos y la criticidad de cada componente para así dimensionar correctamente las actividades de cualificación requeridas.
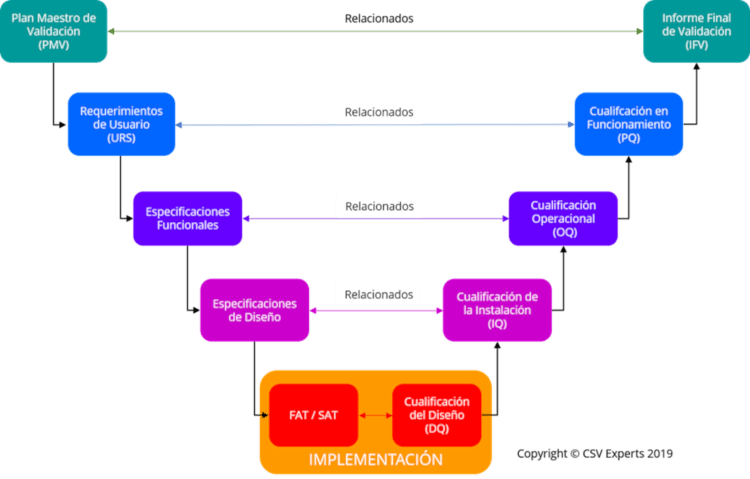
En este sentido, es necesario definir el ciclo de vida de las etapas de cualificación para los equipos. Estas etapas deberán quedar recogidas en el Plan Maestro de Validación (PMV). Este Plan incluye la metodología que se debe seguir para validar los sistemas informatizados de una empresa y todos los aspectos necesarios para llevar a cabo una validación. Incluye, además, un listado de todos los sistemas que se tienen que validar.
Ciclo de vida de los equipos en entornos regulados
Las etapas que comprenden la cualificación de los equipos (ver Anexo 15 de las GMPs) son las siguientes:
Requerimientos de Usuario (URS)
Se deberán definir todos los requerimientos de funcionalidad, de seguridad y regulatorios que deben cumplir los equipos, ya que estos serán la base de las pruebas de cualificación a ejecutar en futuras etapas.
Análisis de Riesgos (RA)
El análisis de riesgos determina el nivel de riesgo de cada uno de los requerimientos especificados en el documento de URS, incluso los requerimientos regulatorios, con el fin de poder dimensionar las pruebas necesarias para mitigar los riesgos detectados en las etapas de IQ/OQ y PQ.
Cualificación del Diseño (DQ)
Esta primera etapa de cualificación, consiste en obtener verificación documentada de que los equipos cumplirán con el propósito para el que están diseñados y sus requisitos están cubiertos por el diseño. Para ello, se trazarán los requerimientos de usuario definidos con la documentación técnica aportada por el proveedor del equipo (Functional Specification, Design Specification, Manuales, etc..).
FactoryAcceptanceTesting (FAT) / Site Acceptance Testing (SAT)
Tanto las pruebas FAT como las pruebas SAT las deberá realizar el proveedor del equipo. Las pruebas FAT se realizarán en las instalaciones del proveedor, y una vez finalizadas y documentadas de forma correcta, se procede al transporte e instalación del equipo en su ubicación final.
Asimismo, en función del componente, el proveedor realiza unas pruebas SAT o commissioning o puesta en marcha del equipo una vez instalado en planta.
Cualificación de la Instalación (IQ)
La cualificación de la instalación del equipo tiene como objetivo verificar la instalación de cada componente atendiendo a los siguientes aspectos:
- Correcta instalación de los componentes de acuerdo a las especificaciones.
- Verificación de los planos y esquemas.
- Existencia de procedimientos de uso, mantenimiento y limpieza.
- Idoneidad de materiales en contacto con el producto.
- Calibración de instrumentos.
Cualificación Operacional (OQ)
Una vez finalizada la cualificación de la instalación, se continua con la cualificación operacional. Esta cualificación está diseñada para verificar el cumplimiento de las funcionalidades de cada equipo atendiendo a:
- Operaciones del equipo.
- Pruebas de comportamiento del equipo en sus límites de operación superiores e inferiores, así como en las peores condiciones de trabajo que se puedan dar (tolerancias).
Cualificación en Funcionamiento (PQ)
Como última etapa de la cualificación, se aborda la cualificación en funcionamiento, la cuál consiste en una monitorización de los equipos en un entorno de producción, para demostrar que el equipo es adecuado para el uso previsto por parte de los usuarios finales, que han recibido la formación adecuada y acorde a su responsabilidad. En esta etapa también se asegura que los procedimientos contienen la información idónea para trabajar con los equipos.
Una vez finalizadas todas las etapas de cualificación, se elaborará el Informe Final de Validación (IFV), donde se demostrará que todas las actividades descritas en el Plan de Validación se han llevado a cabo con éxito. También se incluirán los resultados obtenidos en cada etapa de cualificación y las desviaciones que hayan podido surgir durante el desarrollo de dichas etapas.
Revisión Periódica
Además, será necesario establecer un período adecuado para comprobar que los equipos continúan cualificados a lo largo del tiempo. También será necesario establecer que si se producen cambios, estos se realizan de manera controlada, igual como las actividades que se deben llevar a cabo en una recualificación de los equipos.
La revisión periódica de los sistemas debe realizarse de acuerdo con un procedimiento concreto y se debe documentar incluyendo las acciones correctivas y su seguimiento (plan de acción) hasta su resolución.
La periodicidad de la revisión puede indicarse en el inventario de sistemas informatizados requerido por la normativa que, entre otros datos, puede contener la siguiente información: funciones básicas del sistema, criticidad GxP, categoría GAMP, estado de la validación, frecuencia revisión periódica, responsabilidades sobre el sistema (system owner, process owner, data owner), usos de firma electrónica.
Todo este proceso de validación es un requerimiento de calidad que deben cumplir las empresas del sector farmacéutico, clínico y de dispositivos médicos como parte de las Buenas Prácticas de Fabricación. Los diferentes procesos pasan por una evaluación, documentación y detección de errores existentes para darles una solución.
De este modo se garantiza un correcto procedimiento de los productos tanto por el nivel de calidad exigido como también por la garantía de seguridad que requiere la normativa.
En este sentido, cuando un sistema está validado se entiende que es estable, capaz y robusto, logrando seguridad y optimización del proceso, calidad en el resultado, reducción de costes y aumento de la productividad, además de cumplir con las regulaciones y normas requeridas.
AMBIT BST
En AMBIT somos expertos desde hace más de 15 años en el desarrollo de estrategias y soluciones IT para tu compañía. Somos consultores e integradores en múltiples ámbitos, y si quieres conocer más de las soluciones que te podemos ofrecer, no dudes en ponerte en contacto con nosotros.
Ponemos a disposición de nuestros clientes un equipo técnico con años de experiencia en la realización de este tipo de cualificaciones para el sector farmacéutico. Estamos especializados en liderar la transformación digital del sector salud, farmacéutico y de productos sanitarios para el estricto cumplimiento de las normativas que rigen los entornos GxP.
Si estas interesado en un software cloud que obedezca a estos requisitos, te recomendamos que te descargues este ebook gratuito sobre las ventajas de GxPharma Cloud.








.png)
Cuéntanos tu opinión